Proximo Hi-C Kit for Human Samples
2.300,00 €
Description
Proximity Ligation NGS Library Prep from crude human samples, for Illumina Sequencing.
The Proximo Hi-C method employs cost-effective proximity ligation data generated using in vivo Hi-C to assemble fully phased de novo genomes, detect genetic and epigenetic variation, characterize translocations, copy number variation and TADs, analyze genome and chromatin architecture, study cancer, and advance precision genomics.
• Scaffold chromosome-scale and fully phased human de novo genomes, analyze structural and copy number variants, and perform TAD calling directly from human samples
• No high-molecular-weight DNA extraction required
• User-friendly complete 4-pack Hi-C kit, including NGS library prep reagents
• Short-read compatible; yields libraries for Illumina® sequencing
• Individualized data analysis packages for human de novo genome scaffolding/assembly, haplotype phasing, structural and copy number variant detection, TAD calling and other interaction analyses are available – please contact us to discuss your project details
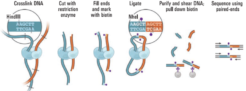
Proximity ligation (Hi-C) libraries are generated from a crude human sample such as tissue, cells, or blood. Interactions are captured by crosslinking, digesting, and creating chimeric junctions that are sequenced. The Hi-C sequence data may be used in conjunction with shotgun/whole genome sequence data obtained by standard NGS approaches for de novo genome assembly and scaffolding, haplotype phasing, detection of structural variants, TAD calling, and other interaction analyses.
Unlock another dimension of the human genome and epigenome with scalable, low-cost NGS to ask and answer new biological questions, and help deliver on the promise of genomics in research and medicine:
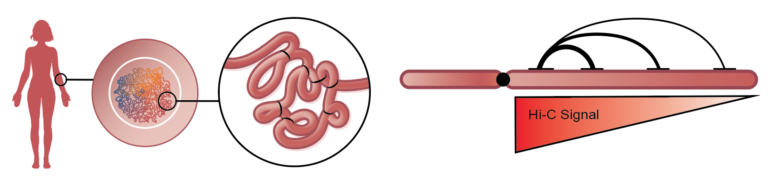
The human genome is an exquisite data set. It encodes every individual’s genetic blueprint in more than three billion base pairs, intricately arranged into 23 chromosome pairs. In addition to the 0.1% difference in DNA sequence between any two individuals, our epigenomes (variation that does not affect DNA sequence) further contribute to our uniqueness. Even though the human genome is the most studied genome of all life on earth, many questions remain unanswered by massively parallel shotgun sequencing. Phase Genomics´ Proximo™ Platform based on Hi-C/proximity ligation utilizes short-read sequencing to map and measure the spatial relationship between any two loci across the full complement of chromosomes in a cell. Computational tools can extract sequential information from this “long-range, short-read” data to enable high-quality de novo genome assembly, phase haplotypes and detect genetic and epigenetic variation, including both small- and large-scale structural variants. Structural variants (SV) are genomic alterations that involve DNA segments larger than 1 kilobase (kb). Examples of SVs include insertions, deletions, inversions, duplications, translocations, copy-number variants (CNVs), as well as variation in chromatin structure conferred by histone modifications. Structural variation comprises millions of bases of heterogeneity within every genome, and is now recognized as one of the primary contributors to the genetic and phenotypic variation that underlies human diversity and disease susceptibility.
Because of their nature, SVs are difficult to study with confidence using short-read sequencing technologies. To unravel these mysteries of our genetic blueprints, we are increasingly relying on emerging methods that provide long-range sequencing information. Hi-C is one of a suite of chromosome conformation capture (or 3C) techniques devised to study the spatial organization of chromatin. Hi-C employs high-throughput, short-read sequencing to identify the nucleotide sequences of genomic loci that are co-located in three-dimensional space, but may be separated by significant distances in the linear genome.
Proximo Workflow:
The proximity ligation (Hi-C) library prep reagents provide a streamlined protocol that does not require extraction of high-molecular weight (HMW) DNA. The processing guidelines for different types of human samples ensure robust results. The 8-step library prep workflow can be completed in 2 working days, and requires only 3 hours of hands-on time. The protocol contains several convenient, safe stopping points. Sequencing may be performed on any Illumina® sequencer. The Proximo Kit yields a dual-indexed proximity ligation library, for which ~50 to 100 million paired-end reads are required. For analyzing your Hi-C sequence data, you also need draft genome sequence data of your samples. If a draft assembly is not yet available for your sample, we can help facilitate traditional sequencing projects prior to Hi-C scaffolding. Contact us for recommendations or to discuss a service project if you do not have prior experience with whole-genome sequencing and assembly.
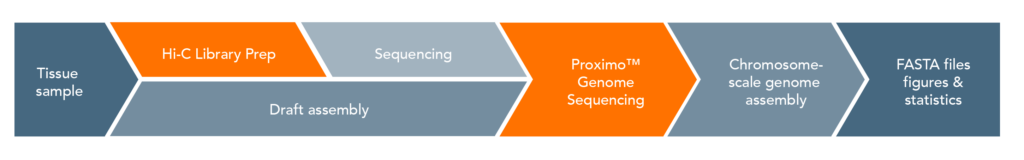
Data analysis is offered optionally as a customized service. E.g. Phase Genomics´ Proximo Genome Scaffolding platform utilizes the three-stage Proximo genome scaffolding algorithm, and Hi-C Proximo data may also be integrated with other computational approaches (e.g. FALCON-Phase), to enable the production of “platinum” (chromosome-scale diploid assembly) human genomes. The open source software FALCON-Phase combines Proximo data with the high accuracy, long-read sequencing data from PacBio®, enabling researchers to create haplotype-resolved genome sequences on a chromosomal scale, even without having parental genome data (see Nature Communications, April 21, 2021).
Other Proximo bioinformatics analysis packages enable topologically associating domain (TAD) calling, as well as the comprehensive analysis of all structural variants (SVs) and copy number variants (CNVs) in a genome.
Please contact us to let us know your project details and receive a quote.