Long-Read NGS

TELL-Seq Barcode Linked-Reads: Long-Read Sequencing for All
• Accurate Long Reads (>100 bp) on Illumina Platform
• Only commercially available barcode linked-reads reagent platform
• Easy scalable 3 hours protocol, all in one PCR tube
• Kits are applicable to any species, genome and project size
• Extremely low input amounts – nanograms of HMW genomic DNA
• Protocols for amplicon– and exome capture– based NGS also available
• Free software package for data analysis
Long-range sequencing information is required for haplotype phasing, de novo assembly and structural variation detection. Current long-read sequencing technologies can provide valuable long-range information but at a high cost with low accuracy and high DNA input requirement.
Our partner Universal Sequencing Technology have developed the single-tube Transposase Enzyme Linked Long-read Sequencing (TELL-seqTM) technology ( Chen et al., Genome Research, June 2020 ), which enables a low-cost, high-accuracy and high-throughput short-read second-generation sequencer to generate over 100 kb long-range sequencing information with as little as 0.1 ng input material. And all of this without the requirement for any additional costly devices.
The TELL-Seq Barcode Linked-Reads Solution:
In a PCR tube, millions of clonally barcoded beads are used to uniquely barcode long DNA molecules in an open bulk reaction without dilution and compartmentation. The barcode linked reads are used to successfully assemble genomes ranging from microbes to human. These linked-reads also generate mega-base-long phased blocks for haplotyping and provide a cost-effective tool for detecting structural variants in a genome, which are important to identify compound heterozygosity in recessive Mendelian diseases and discover genetic drivers and diagnostic biomarkers in cancers.
(A)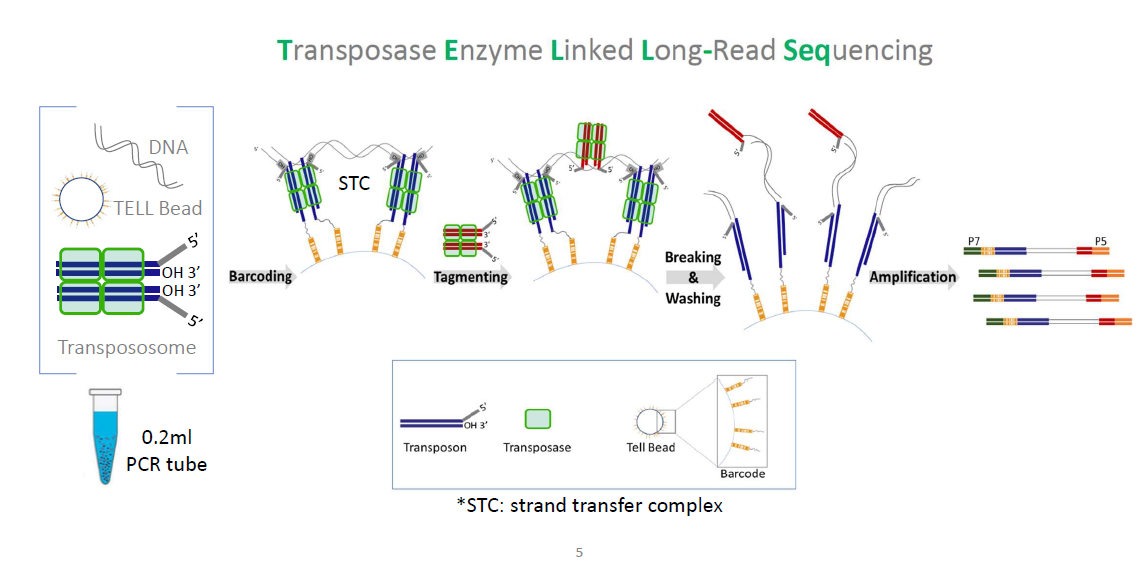
(B)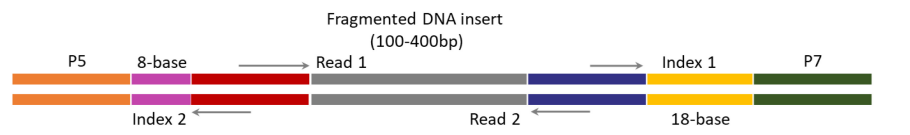
(A) Diagram of TELL-seq library preparation procedure. In a 0.2-mL PCR tube, 0.1 ng to 5 ng genomic DNA was mixed with 3–10 million barcoded TELL beads and transpososomes for the clonal barcoding reaction. Genomic DNA fragments were captured on the barcoded TELL beads via connecting strand transfer complexes (STCs) to barcode oligos on the bead surface. A tagging between STCs by a second transpososome introduced a second priming site for library amplification. After breaking the STCs and washing the magnetic TELL beads, sequencing library molecules were amplified off beads with P5 and P7 adaptor sequences incorporated at the same time. The total library procedure took ∼3 h.
(B) TELL-seq library structure for Illumina sequencing systems. Index 1 comprises 18-base TELL-seq molecular barcode; Index 2 comprises 8-base barcode for sample indexing.
See also UST´s whitepaper “A Brief Introduction to Linked-Read Technology”
User Manuals for TELL-Seq Kits and Reagents
TELL-Seq Data Analysis Software – Free Download
Applications:
• ![]() De novo Genome Assembly and Scaffolding
De novo Genome Assembly and Scaffolding
Using the TELL-Seq™ WGS library prep kits you can sequence novel genomes of any size – small bacterial genomes, medium sized insect, plant, and fungi genomes, and large animal and plant genomes. If you really want to understand a new genome, including gene synteny, you must look at long-reads. The TELL-Seq™ kit will turn an ordinary short-read 2nd generation DNA sequencer into a long-read 3rd generation DNA sequencer. The TELL-Seq Microbial WGS Library Kits are specifically designed for samples with less than 50Mb genome size to prepare TELL-Seq™ libraries with the least cost. For genomes > 50Mb we offer the TELL-Seq WGS Library Kits.
Examples:
9 previously unsequenced insect genomes were successfully assembled in a pilot study of the Earth BioGenome Project using TELL-Seq at the University of Illinois. Attend Illumina´s webinar to learn more.
Draft genome assemblies of the avian louse Brueelia nebulosa and its associates using long-read sequencing from an individual specimen
Genome Sequence of Lichtheimia ornata, an Emerging Opportunistic Mucorales Pathogen
![]()
• Metagenomics: Decipher microbial genomes in complex mixed samples
Samples containing disparate microbes are essential to gaining insight into our ecosystem as well as human disease. The TELL-Seq™ WGS Library Prep kit will allow you to sequence samples at a metagenomic scale, providing long-read like results with the ease of short-read sequencing. To see what is truly in a mixed microbial sample use the power of long-read DNA sequencing with the simplicity of short-read sequencing.
Examples:
Characterizing the microbial metagenome of calcareous stromatolite formations in the San Felipe Creek in Anza Borrego Desert (2023)
Illumina Application Note “Microbial de novo assembly with linked-read technology”
![]() • Structural Variation
• Structural Variation
Short-read DNA sequencers have trouble identifying large structural variants such as copy number variations (CNVs), translocations, duplications, deletions, insertions, and inversions. The TELL-Seq™ WGS Library Prep kit links short-reads turning them into long-reads. The linked, long-read data that you get using the TELL-Seq™ kit will allow you to take a step back and see what you were missing.
Examples:
Human commensal Candida albicans strains demonstrate substantial within-host diversity and retained pathogenic potential demonstrated by TELL-Seq
Resolving of Structural Variant Discrepancies in Human GIAB Samples by TELL-Seq
Whole-genome risk prediction of common diseases in human preimplantation embryos
Linked‑read sequencing for detecting short tandem repeat expansions
![]() • Whole Genome and Targeted Haplotype Phasing
• Whole Genome and Targeted Haplotype Phasing
Knowing which homologous chromosome has which sequence is essential to understanding a genome. Phasing provides haplotype information which is important for understanding complex traits and variant linkages. However, Whole Genome Phasing is difficult with 2nd generation, short-read, DNA sequencing technology. The TELL-Seq™ Library Prep Kit helps with this by linking short-reads to larger fragments and generate long-range information for both whole genome sequencing or targeted phasing applications.
Examples:
SpLitteR: Diploid genome assembly using TELL-Seq linked-reads and assembly graphs
Illumina Application Note: “UST TELL-Seq using Illumina NGS platforms enables genome-scale haplotype”
OVERVIEW: TELL-Seq Publications and Data
Visit the FAQ site for detailed technical information about TELL-Seq
Please also visit our Hi-C Sequencing website.
Showing all 16 results